-
Belite Bio Reports Third Quarter 2023 Operational Highlights and Financial Results
Источник: Nasdaq GlobeNewswire / 13 ноя 2023 20:15:47 America/New_York
- Completed enrollment in pivotal Phase 3 “DRAGON” trial for Tinlarebant in adolescent Stargardt disease (“STGD1”) with 104 subjects enrolled across 11 countries worldwide
- First subject dosed with Tinlarebant in pivotal global Phase 3 “PHOENIX” trial in Geographic Atrophy (“GA”)
- Oral, once-daily Tinlarebant continues to be safe and well tolerated, slowing expansion of autofluorescence, reducing incident atrophic retinal lesion growth rate, and stabilizing visual acuity up to 24-months in Phase 2 STGD1 trial (“LBS-008-CT02”)
- Interim Phase 3 safety and efficacy data from pivotal “DRAGON” trial expected in 2H 2024
- Conference Call and Webcast Tuesday, November 14, 2023, at 4:30 p.m. ET
SAN DIEGO, Nov. 13, 2023 (GLOBE NEWSWIRE) -- Belite Bio, Inc (NASDAQ: BLTE) (“Belite” or the “Company”), a clinical-stage biopharmaceutical drug development company focused on advancing novel therapeutics targeting degenerative retinal diseases that have significant unmet medical needs, today announced its financial results for the three-months ended September 30, 2023, and provided a general business update.
“We made meaningful progress in the quarter advancing our clinical trials for Tinlarebant and are excited by our results,” said Dr. Tom Lin, Chairman and CEO of Belite Bio. “Results from our Phase 2 trial of Tinlarebant in childhood-onset Stargardt Disease showed Tinlarebant lowered retinal lesion growth versus patients in a 24-month natural history study and visual acuity was stabilized, all while remaining safe and well-tolerated. Additionally, in the quarter, we completed enrollment for the DRAGON trial, dosed our first subject and made substantial progress in the PHOENIX trial. We remain focused on our vision to bring novel therapies to patients suffering from degenerative retinal diseases.”
Professor John Grigg, Head Specialty of Ophthalmology at the University of Sydney and Consultant Ophthalmologist at the Sydney Children’s Hospitals Network at Westmead and Sydney Eye Hospital, presented the results of the Phase 2 trial of Tinlarebant in adolescent Stargardt disease patients at the American Association of Ophthalmology annual meeting early this month and commented, “The results of the Phase 2 study with 24-month treatment of Tinlarebant in adolescents were promising. Stargardt is a debilitating disease that may have a life-changing impact on those diagnosed with the disease. The disease most often progresses quickly, and vision deteriorates rapidly at a very young age. The Phase 2 data continued to demonstrate slowing of the disease progression in the study cohort and the stabilization in several structural and functional parameters including stabilization of visual acuity. We are highly encouraged by these results and the potential to impact the lives of patients suffering from a disease for which there currently is no FDA-approved treatment.”
Third Quarter 2023 Business Highlights and Upcoming Milestones:
Clinical Highlights
Tinlarebant (LBS-008) is designed to be an oral, potent, once-daily retinol binding protein 4 (RBP4) antagonist that decreases RBP4 levels in the blood and reduces vitamin A (retinol) delivery to the eye without disrupting systemic retinol delivery to other tissues. Vitamin A is critical to normal vision but can accumulate as toxic byproducts leading to retinal cell death and vision loss diseases such as STGD1 and GA, the advanced form of dry Age-Related Macular Degeneration (dry AMD).
- Stargardt disease (STGD1): Accumulation of cytotoxic bisretinoids has been implicated in the onset and progression of STGD1. Tinlarebant has been granted Fast Track and Rare Pediatric Disease (RPD) designations by the U.S. Food and Drug Administration (FDA), and orphan drug designation (ODD) in the U.S. and Europe for STGD1. There are currently no FDA-approved treatments for STGD1.
- LBS-008-CT02 trial: Ongoing, open-label, 2-year Phase 1b/2 trial in adolescent STGD1 subjects
- A total of 12 adolescent STGD1 subjects aged 12-18 years completed 24-months of treatment in the Phase 2 study of Tinlarebant. Key study findings are as follows:
- Tinlarebant was safe and well-tolerated with no withdrawals due to adverse events.
- Retinal imaging showed that 5 of 12 subjects remained free of atrophic retinal lesions (referred to as definitely decreased autofluorescence or DDAF) after 24 months of Tinlarebant treatment.
- A comparison of the 24-month DDAF lesion growth between Tinlarebant-treated subjects and ProgStar participants possessing similar baseline characteristics (aged ≤18 years) showed a sustained lower DDAF lesion growth in Tinlarebant-treated subjects over the 24-month treatment period (p<0.001).
- Visual acuity was stabilized in the majority of subjects during the study with a mean loss of five letters following 24 months of treatment (a loss of <10 letters is not considered clinically significant).
- Tinlarebant was safe and well-tolerated with no withdrawals due to adverse events.
- A total of 12 adolescent STGD1 subjects aged 12-18 years completed 24-months of treatment in the Phase 2 study of Tinlarebant. Key study findings are as follows:
- LBS-008-CT02 trial: Ongoing, open-label, 2-year Phase 1b/2 trial in adolescent STGD1 subjects
*Only 50 patients from ProgStar Cohort (aged ≤18 ) were included in the analysis due to one subject having ungradable screening FAF data.1. Strauss RW, Ho A, Muñoz B, et al. ProgStar Report No. 1. Ophthalmology. 2016;123(4):817-28.
2. Strauss RW, Muñoz B, Ho A, et al. ProgStar Report No. 9. JAMA Ophthalmol. 2017; 135(11):1232-1241.- Pivotal DRAGON trial: 2-year, randomized, double-masked, placebo-controlled, global, multi-center, pivotal Phase 3 trial in STGD1 subjects aged 12-20 years old:
- Enrollment complete, with 104 subjects across 11 countries worldwide including sites in the U.S., the United Kingdom, Germany, Belgium, France, Switzerland, China, Hong Kong, Taiwan, Australia, and the Netherlands.
- Primary efficacy endpoint is slowing of lesion growth rate; safety and tolerability will also be assessed.
- 2H 2024: Interim efficacy and safety data expected.
- Enrollment complete, with 104 subjects across 11 countries worldwide including sites in the U.S., the United Kingdom, Germany, Belgium, France, Switzerland, China, Hong Kong, Taiwan, Australia, and the Netherlands.
Geographic Atrophy (GA): GA, the advanced form of dry AMD, is a chronic degenerative disease of the retina that leads to blindness in the elderly. Accumulation of toxic vitamin A byproducts (bisretinoids) has been implicated in the progression of GA. There are currently no FDA-approved orally administered treatments for GA.
- Pivotal PHOENIX Trial: 2-year prospective, randomized (2:1, active:placebo, n ~430), double-masked, placebo-controlled, global, multi-center, Phase 3 trial in subjects with GA.
- Primary efficacy endpoint is slowing of lesion growth rate; safety and tolerability will also be assessed.
- First subject has been dosed.
- Interim analysis expected at mid-point of the trial.
- Primary efficacy endpoint is slowing of lesion growth rate; safety and tolerability will also be assessed.
Corporate Highlights
- For the three months ended September 30, 2023, the Company strengthened its balance sheet with $5.0 million of gross proceeds from the exercise of warrants granted in the underwritten follow-on offering in May 2023.
- For the three months ended September 30, 2023, the Company raised $0.64 million of net proceeds from its at-the-market offering program established in June 2023.
Risks and Uncertainties
Based on current business plans and financial expectations, the Company expects that it will be a “passive foreign investment company” within the meaning of Section 1297 of the U.S. Internal Revenue Code of 1986, as amended (“PFIC”) for its current tax year 2023, and may be a PFIC in one or more future tax years, which may have adverse U.S. federal income tax consequences for U.S. securityholders.
If the Company is a PFIC for any year during a U.S. taxpayer’s holding period of the Company’s ADSs or ordinary shares, then such U.S. taxpayer generally will be required to treat any gain realized upon a disposition of the Company’s ADSs, ordinary shares or warrants or any so-called “excess distribution” received on such ADSs, ordinary shares or warrants as ordinary income, and to pay an interest charge on a portion of such gain or distribution. In certain circumstances, the sum of the tax and the interest charge may exceed the total amount of proceeds realized on the disposition, or the amount of excess distribution received, by the U.S. taxpayer. Subject to certain limitations, these tax consequences may be mitigated if a U.S. taxpayer makes a timely and effective “qualified electing fund” (“QEF”) election within the meaning of Section 1295 of the Code (a “QEF Election”) or a “mark-to-market election” within the meaning of Section 1296 of the Code (a “Mark-to-Market Election”). The QEF Election is not available with respect to warrants until those warrants are exercised. Each investor in a warrant (and, in particular, any such investor that holds warrants but not ADSs or ordinary shares) who is a U.S. taxpayer should consult its own tax advisor regarding the tax consequences of the PFIC rules and the ownership and disposition of the warrants. The Mark-to-Market Election is only available with respect to ADSs, ordinary shares or warrants regularly traded on a qualified exchange or market. Subject to certain additional limitations, such elections may be made with respect to the Company’s ADSs or ordinary shares (and with respect to the Mark-to-Market Election, the warrants). A U.S. taxpayer who makes a timely and effective QEF Election generally must report on a current basis its share of the Company’s net capital gain and ordinary earnings for any year in which the Company is a PFIC, whether or not the Company distributes any amounts with respect to the ADSs or ordinary shares. However, U.S. taxpayers should be aware that there can be no assurance that the Company will satisfy the record keeping requirements that apply to a QEF, or that the Company will supply U.S. taxpayers with information that such U.S. taxpayers require to report under the QEF Election rules, in the event that the Company is a PFIC and a U.S. taxpayer wishes to make a QEF Election. Thus, U.S. taxpayers may not be able to make a QEF Election with respect to their ADSs or ordinary shares. A U.S. taxpayer who makes the Mark-to-Market Election generally must include as ordinary income each year the excess of the fair market value of the ADSs or ordinary shares over the taxpayer’s basis therein. Each potential investor who is a U.S. taxpayer should consult its own tax advisor regarding the tax consequences of the PFIC rules and the ownership and disposition of ADSs or ordinary shares, including the availability of and procedure for making a QEF Election or a Mark-to-Market Election.
Third Quarter 2023 Financial Results:
Cash: As of September 30, 2023, the Company had $54.5 million in cash.
R&D Expenses:
For the three months ended September 30, 2023, research and development expenses were $8.7 million compared to $1.2 million for the same period in 2022. The increase resulted primarily from increases in (x) expenses for conducting the DRAGON and PHOENIX trials and (y) wages and salaries due to share-based compensation granted to our R&D team in the third quarter of 2023. For the nine months ended September 30, 2023, research and development expenses were $20.0 million compared to $3.6 million for the same period in 2022. The increase in research and development expenses was primarily attributable to increases in (i) expenses related to conducting the DRAGON and PHOENIX trials, and (ii) wages and salaries due to our R&D team expansion and share-based compensation granted in the third quarter of 2023.
G&A Expenses:
For the three months ended September 30, 2023, general and administration expenses were $2.2 million compared to $1.4 million for the same period in 2022. The increase resulted primarily from an increase in share-based compensation granted in the third quarter of 2023. For the nine months ended September 30, 2023, general and administration expenses were $4.7 million compared to $2.5 million for the same period in 2022. The increase resulted also primarily from an increase in share-based compensation granted in the third quarter of 2023 and an increase in professional service fees.
Net Loss:
For the three months ended September 30, 2023, the Company reported a net loss of $10.9 million or ($0.40) per share compared to $2.4 million or ($0.10) per share for the same period in 2022. For the nine months ended September 30, 2023, the Company reported a net loss of $24.6 million or ($0.95) per share, compared to a net loss of $5.9 million or ($0.32) per share for the same period in 2022.
Webcast Information
Belite Bio will host a webcast on Tuesday, November 14, 2023, at 4:30 p.m. Eastern Time to discuss the Company’s financial results and provide a business update. To join the webcast, please visit https://wsw.com/webcast/cc/blte/1366794. A replay will be available for approximately 90 days following the event.
About Belite Bio
Belite Bio is a clinical-stage biopharmaceutical drug development company focused on advancing novel therapeutics targeting degenerative retinal diseases with significant unmet medical needs, such as STGD1 and GA in advanced dry AMD, in addition to specific metabolic diseases. For more information, follow us on Twitter, Instagram, LinkedIn, Facebook or visit us at www.belitebio.com.
Important Cautions Regarding Forward Looking Statements
This press release contains forward-looking statements about future expectations, plans and prospects, as well as any other statements regarding matters that are not historical facts. These statements include but are not limited to statements regarding the potential implications of clinical data for patients, clinical development, regulatory milestones of its product candidates, and any other statements containing the words “expect”, “will”, “believe”, and other similar expressions. Actual results may differ materially from those indicated in the forward-looking statements as a result of various important factors, including but not limited to Belite Bio’s ability to demonstrate the safety and efficacy of its drug candidates; the clinical results for its drug candidates, which may not support further development or regulatory approval; expectations for the timing of initiation, enrollment and completion of, and data relating to, its clinical trials; the content and timing of decisions made by the relevant regulatory authorities regarding regulatory approval of Belite Bio’s drug candidates; whether additional clinical trials may be required for DRAGON or PHOENIX studies based on their respective data; the potential efficacy of Tinlarebant, as well as those risks more fully discussed in the “Risk Factors” section in Belite Bio’s filings with the U.S. Securities and Exchange Commission. All forward-looking statements are based on information currently available to Belite Bio, and Belite Bio undertakes no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as may be required by law.
BELITE BIO, INC
UNAUDITED CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(Amounts in thousands of US Dollars, except share and per share amounts)For the Three Months For the Nine Months Ended September 30, Ended September 30, 2022 2023 2022 2023 Expenses Research and development 1,185 8,743 3,643 19,982 General and administrative 1,355 2,218 2,457 4,731 Total operating expenses 2,540 10,961 6,100 24,713 Loss from operations (2,540) (10,961) (6,100) (24,713) Other income (expense): Total other income, net 137 27 236 81 Loss before income tax (2,403) (10,934) (5,864) (24,632) Income tax expense - 1 - 10 Net loss (2,403) (10,935) (5,864) (24,642)) Other comprehensive income (loss) Foreign currency translation adjustments, net of nil tax (166) (55) (323) (115) Total comprehensive loss (2,569) (10,990) (6,187) (24,757) Weighted average number of ordinary shares used in per share calculation: - Basic and Diluted 24,869,038 27,315,550 18,321,088 26,013,012 Net loss per ordinary share - Basic and Diluted $ (0.10) $ (0.40) $ (0.32) $ (0.95) BELITE BIO, INC
UNAUDITED CONDENSED CONSOLIDATED BALANCE SHEETS
(Amounts in thousands of US Dollars, except share amounts)December 31, September 30, 2022 2023 Current assets $ 42,807 $ 56,135 Other assets 1,466 1,463 TOTAL ASSETS $ 44,273 $ 57,598 TOTAL LIABILITIES $ 2,772 $ 4,804 TOTAL SHAREHOLDERS’ EQUITY 41,501 52,794 TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY $ 44,273 $ 57,598 Ordinary shares authorized 492,179,086 400,000,000 Ordinary shares issued and outstanding 24,898,908 27,599,244 Media and Investor Relations Contact:
Jennifer Wu /ir@belitebio.com
Argot Partners /ir@belitebio.comPhotos accompanying this announcement are available at:
https://www.globenewswire.com/NewsRoom/AttachmentNg/a9faecbb-230b-4dd2-8fd7-0796ba383139
https://www.globenewswire.com/NewsRoom/AttachmentNg/4c5b2dd3-8bb5-40c0-96d9-2ec4422ea4fd

Comparison of the 24-month DDAF lesion growth between Tinlarebant-treated subjects and ProgStar participants
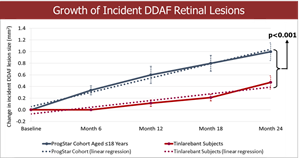
Comparison of the 24-month DDAF lesion growth between Tinlarebant-treated subjects and ProgStar participants
Comparison between Tinlarebant-treated subjects and ProgStar participants
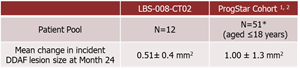
Comparison between Tinlarebant-treated subjects and ProgStar participants
- Completed enrollment in pivotal Phase 3 “DRAGON” trial for Tinlarebant in adolescent Stargardt disease (“STGD1”) with 104 subjects enrolled across 11 countries worldwide